The receptors of the lungs and airways are innervated through the vagi and superior laryngeal and trigeminal nerves, and respond, as in other hollow visceral structures, to irritation of the lining layers and changes in distending forces. The mechanoreceptors associated with the respiratory muscles are innervated by spinal nerves and, like those in other skeletal muscles, monitor changes in joint movement and in the length and tension of the muscle itself.
Pulmonary Receptors
There are basically three types of pulmonary receptors: stretch receptors in the smooth muscles of the airway, irritant receptors in the airway epithelium, and J (juxtacapillary) receptors situated in the lung interstitium.
Stretch Receptors
Stretch receptors are innervated by large myelinated fibers. As the lung is inflated, these receptors inhibit inspiration, promote expiration, and initiate the Hering-Breuer reflex. In animals, lung inflation cuts short inspiration and produces expiratory apnea; the duration of apnea is proportional to the degree of inflation.
Direct measurements of stretch-receptor activity indicate that stretch receptors in humans are excited by even small changes in lung volume during quiet breathing. In humans, however, unlike what occurs in animals, vagal blockade to abolish stretch-receptor input does not affect breathing frequency or tidal volume at rest. Vagal blockade in both humans and animals does, however, prevent the increase in breathing frequency that occurs when ventilation is stimulated by hypercapnia or hypoxia and tidal volume is larger.
In animals, stretch-receptor activity helps to preserve tidal volume whenever the usual movements of the lung are hindered by changes in airways resistance or respiratory system compliance. Anything that retards lung inflation diminishes inspiratory inhibitory stretch-receptor activity. Therefore, inspiration is prolonged and tidal volume tends to approach its usual level when the airway is obstructed or respiratory compliance is reduced, despite mechanical interference. When expiration is hindered and lung deflation slowed, increased stretch-receptor activity heightens the force of contraction of the expiratory muscles and also prolongs expiratory time. Both these stretch-receptor actions tend to prevent mechanical impediments to expiration from increasing end-expiratory volume and, as a consequence, decreasing the resting length of the inspiratory muscle. Stretch-receptor activity, by promoting full expiration, helps preserve inspiratory muscle function.
Although stretch receptors are not important in humans in shaping resting breathing patterns, they may help maintain tidal volume when breathing is stimulated or lung or chest-wall mechanical performance is impaired. The increase in breathing frequency caused by stretch-receptor activity in animals and during stimulated breathing in humans decreases the work of breathing of the respiratory muscles, conserving the energy that has to be expended to produce gas exchange. Although it is well-known that peripheral inputs from lung mechanoreceptors strongly affect the timing of respiratory motor activity, at the present time it is difficult to separate clearly the ventilatory effects of the pulmonary stretch-receptor afferents from those of other vagal sensory components (e.g., irritant and C-fiber afferents). However, changes in vagal afferent activity elicited by phasic lung volume changes seem to control predominantly the duration of inspiration, whereas tonic inputs predominantly affect the duration of expiration.
Irritant Receptors
Irritant receptors, like the stretch receptors, are innervated by myelinated fibers, whereas unmyelinated fibers supply the J receptors. Unlike the stretch receptors, both irritant and J receptors are rapid-adapting (within seconds). Neither irritant nor J receptors have a pattern of firing that is related to the phases of inspiration and expiration. Consequently, it is believed that neither receptor has an important influence in determining the pattern of breathing at rest.
Mechanical stimulation of the airways or the inhalation of potentially noxious agents (e.g., particulate matter, nitrogen dioxide, sulfur dioxide, ammonia, and antigens) seems to excite irritant receptors and produce airway constriction. Stimulation of irritant receptors augments the activity of the inspiratory neurons and, by interaction with the stretch receptors, promotes rapid, shallow breathing. This pattern of breathing, in combination with airway constriction, may limit penetration of dangerous agents into the lung and prevent them from reacting with the gas-exchanging surfaces.
The inspiratory augmenting effect of irritant-receptor excitation and the increase in breathing frequency it produces may help maintain ventilation in asthmatic patients, even when the work of breathing is massively increased.
Irritant receptors can be excited by traction on the airways and are stimulated if atelectasis reduces lung compliance. These receptors seem to cause augmented breathing and the large sighs that occur sporadically during normal breathing, and help to open collapsed areas of the lung. As a consequence, irritant receptors help maintain adequate gas exchange.
J Receptors
J receptors are stimulated by pulmonary interstitial edema, but they also can be activated by various chemical agents, such as histamine, halothane, and phenyldiguanide. Activation of the J receptors causes laryngeal closure and apnea, followed by rapid, shallow breathing. When pulmonary edema develops as a result of exercise, J receptors seem to depress the activity of the exercising limbs by a somatic reflex involving cingulate gyrus. J receptors, together with irritant receptors, may be responsible for the tachypnea seen in patients with pulmonary embolus, pulmonary edema, and pneumonia.
Laryngeal Receptors
Mechanoreceptors and chemoreceptors in the upper airway reflexively affect the level and pattern of breathing, motor outflow to the upper airway and chest-wall muscles, and airway tone. The best-studied of the upper airway receptors are the laryngeal receptors. In fact, all areas of the laryngeal mucosa and deeper structures contain sensory nerve endings. Several types of laryngeal receptors have been described: (1) pressure receptors, (2) “drive” receptors, and (3) cold receptors.
Pressure receptors, the most numerous of the laryngeal receptors, are activated by increases in negative (intraluminal less than extraluminal pressure) or positive transmural pressure. Pressure receptors fire in response to both dynamic and static pressure changes, and are slow-adapting. Approximately, two thirds of the pressure receptors respond to negative pressure; the remaining third respond to positive pressure. Approximately one half of laryngeal pressure receptors demonstrate a respiratory modulation in the absence of air flow in the isolated, bypassed upper airway, suggesting that they respond to laryngeal muscle shortening in response to descending motor drive. These so-called drive receptors fire primarily during inspiration. Their firing pattern is diminished by paralysis of the intrinsic muscles of the larynx.
Reflexes elicited by laryngeal pressure receptors tend to stabilize the upper airway, retard its tendency to collapse in response to subatmospheric pressure, and re-establish its patency following occlusion. Laryngeal pressure receptors reflexively activate upper airway muscles while inhibiting inspiratory muscles of the chest wall. Negative transmural airway pressure reflexes increase the activity of inspiratory upper airway muscles (e.g., genioglossus, sternohyoid, cricothyroid, levator alae nasi, posterior arytenoids), advance the onset of the upper airway-muscle EMG relative to that of the diaphragm, increase the duration of inspiration and expiration, and decrease the average rate of rise of diaphragmatic and inspiratory intercostal EMG activity. (Normally, activation of upper airway muscles occurs 50 to 100 ms before the outset of diaphragmatic activation.) Reflex responses to negative pressure in the upper airway mediated by pressure receptors may explain the greater tidal volume, expiratory time, and ventilation during nasal than in tracheostomy breathing in conscious animals and humans.
In contrast to pressure receptors, laryngeal cold receptors are silent near body temperature but are activated by decreases in laryngeal temperature to 34°C or below. When active, cold receptors demonstrate a phasic, inspiratory firing pattern and, in contrast to pressure receptors, appear to adapt rapidly. Cold receptors appear to be located superficially in the mucosa on the edge of the vocal cords near the arytenoid process. Increases in lower airway resistance elicited by laryngeal cooling may be mediated by these receptors.
Finally, mechanical or chemical irritation of the larynx (e.g., probe contact or application of acid) elicits cough, laryngeal closure, bronchoconstriction, an increase in tracheal production of mucus, and a decrease in heart rate and blood pressure. These reflex responses to laryngeal irritation suggest that laryngeal chemoreceptors and mechanoreceptors function to protect the lower airway from aspiration or inhalation of toxic fumes.
Of interest, reflex responses to laryngeal stimulation appear to be state-dependent and are qualitatively different during wakefulness and sleep. For example, in the dog, application of distilled water to the larynx during wakefulness consistently elicits cough and bronchoconstriction. In contrast, the same maneuver performed during REM sleep does not stimulate cough, but rather elicits apnea and bradycardia.
Chest-Wall Receptors
Three types of receptors in the chest wall—joint, tendon, and spindle receptors—signal changes in the force exerted by the respiratory muscles and movement of the chest wall. Specialized Ruffini receptors, as well as pacinian and Golgi organs, are present in joints. Joint-receptor activity, which can be consciously perceived, varies with the degree and rate of change of rib movement.
Inputs arising from muscular receptors, both proprioceptive (particularly muscle spindle) and nociceptor afferent (types III and IV) endings, influence the level and timing of respiratory activity. Proprioceptor afferents (chiefly from the intercostal and abdominal muscles) project to the phrenic motor neurons, where their effect is on firing rate only, and to medullary respiratory neurons in the DRG and NRA, where their predominant effect is on respiratory timing.
Tendon organs in the intercostal muscles and diaphragm monitor the force of muscle contraction and produce an inspiratory inhibitory effect. It was once thought that tendon organ activity was provoked only by unusual levels of muscle force, but it is now believed that tendon organs are stimulated by even small changes in force. Tendon organ input may be important in regulating both intercostal muscle and diaphragmatic contraction during breathing at rest.
Muscle spindles, which are abundant in the intercostal muscles but scarce in the diaphragm, are involved in several kinds of intercostal respiratory reflexes and also help coordinate breathing during changes in posture and speech.
Figure 7 shows schematically the operation of the spindle and its neural connections. Spindles are located on intrafusal muscle fibers aligned in parallel with extrafusal fibers, which move the ribs. Motor innervation of the extrafusal fibers originates in alpha motor neurons, whereas the intrafusal fibers receive motor innervation from gamma (fusimotor) motor neurons. Passive stretch of an intercostal spindle by lateral flexion of the trunk, for example, increases spindle afferent activity and activates a monosynaptic segmental reflex that causes contraction of the parent extrafusal fiber and restores the upright position. The spindles also can be stretched by an efferent fusimotor discharge, which causes contraction and shortening of the intrafusal fiber itself. Some fusimotor fibers fire phasically, so that their rate of discharge rises during inspiration and falls during expiration; other fusimotor fibers are tonically active. The cerebellum determines the balance between tonically and phasically active fusimotor fibers. Without phasic fusimotor activity, spindle discharge would decrease when the extrafusal fibers contract during inspiration. Simultaneous activation of fusimotor and alpha motor neurons causes the spindles to be under continuous stretch during inspiration and enhances the contribution made by the intercostal muscles to respiration. If inspiratory movements are impeded, afferent activity from a spindle innervated by a phasically active fusimotor fiber is enhanced, thus increasing inspiratory muscle force and helping to preserve tidal volume. Activity from lower intercostal muscle spindles, through an intersegmental spinal reflex, also enhances diaphragmatic contraction, allowing the diaphragm to contribute to the compensatory increase in muscle force that occurs when respiratory movements are hindered. In contrast, stretch of the intercostal spindles in the midthoracic region of the chest decreases the duration of inspiration and diminishes the force of inspiratory muscle contraction. This reflex may cut short ineffective inspirations. Ineffective inspirations sometimes are seen in the newborn when the negative intrathoracic pressure produced by powerful diaphragmatic contraction causes paradoxical inward movement of the flexible infant rib cage.
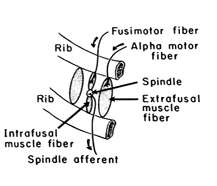
FIG. 7. Intercostal muscle spindle.
Of considerable importance, spindle afferents reach the highest level of the central nervous system, the sensorimotor cortex. Projection of spindle afferent activity to the cerebral cortex allows respiratory muscle length and tension to be sensed consciously and modulated with great precision, thereby allowing complex volitional acts to be performed (e.g., speaking, playing a wind instrument). Spindle afferent activity also likely contributes to the sense of breathlessness. It has been suggested that dyspnea occurs when spindle afferent activity is “high” relative to the intensity of central motor activity to the inspiratory muscles. This concept, which has been termed length-tension inappropriateness, explains the dyspnea that arises in the setting of lung diseases that increase inspiratory muscle load and impede muscle shortening. Of interest, the sense of breathlessness can be affected in patients with chronic obstructive pulmonary disease (COPD) by application of vibratory stimuli to the intercostal muscles, which changes spindle afferent activity. Dyspnea is ameliorated by vibratory stimuli applied in phase with muscle contraction and worsened when the vibratory stimulus is applied out of phase with muscle contraction.
Integration of Afferent Input
Although it is clear that afferent input to the medullary respiratory neurons from mechanoreceptors in the lungs, respiratory muscles, and cardiovascular and thermal regulatory systems (and even the exercising limbs) have significant effects on breathing, the precise manner in which these inputs are integrated is poorly understood. However, the changes in respiratory motor activity elicited by changes in these inputs are not stereotyped. The reflex responses to these inputs may affect the motor output to some respiratory muscles more than others. Pulmonary stretch-receptor input inhibits chest-wall muscle activity (i.e., diaphragm and external intercostal muscles) but increases the activity of the upper airway-dilating muscles (i.e., posterior cricoarytenoid) and the chest-wall expiratory muscles. Even more interesting, some receptors seem to have opposing effects on muscles that normally act as agonists. For example, stimulation of esophageal mechanoreceptors by balloon distension of the distal esophagus reflexively inhibits diaphragmatic activity, both costal and vertebral, but enhances external intercostal activity.












